看完諾貝爾化學獎,我開始幻想ChatGPT能拿文學獎
如果說10月8日頒發的諾貝爾物理學獎是與人工智能有點曖昧不清,那麼10月9日揭曉的諾貝爾化學獎就徹底“不裝了”,看來ChatGPT拿文學獎也指日可待了(bushi)。
2024年的諾貝爾化學獎授予大衛·貝克爾(David Baker),以表彰其計算蛋白質設計方面的研究;以及德米斯·哈薩比斯(Demis Hassabis)和約翰·詹珀(John Jumper),以表彰其在蛋白質結構預測方面的研究。

這兩項研究都與人工智能與生物學的深度結合有關。蛋白質設計和蛋白質結構預測方面的工作,若非人工智能的加持,註定是一個極其有意義但同時極其無解的領域。
下面,我們就來一起了解一下,爲什麼這項工作意義重大而又如此困難,人工智能又爲解決這一問題作出了怎樣的貢獻。
蛋白質的功能與結構
蛋白質是一切生命活動的體現者。大腦傳遞信息,靠的是蛋白質搬運電荷;肌肉收縮,靠的是蛋白質互相拔河;細胞需要能量,靠的是蛋白質運輸氧氣。癌症、阿茲海默症、艾滋病、糖尿病……幾乎任何疾病都與蛋白質有着千絲萬縷的聯繫。
換句話說,想要研究透徹生命,就必須研究透徹蛋白質;想要研究透徹蛋白質,就必須完全瞭解其結構的祕密。
很久之前人們就知道,蛋白質雖然種類可能上億,形態結構千變萬化,但是它們大部分都是由20種組成元件拼成的,這些元件就是氨基酸。就像樂高積木雖然能拼出形形色色的物體,但是其基礎小塊的數量卻很少。
一個蛋白質到底選用了哪些氨基酸以及它們按順序排列成多肽鏈,這被稱爲蛋白質的一級結構;這些氨基酸排好序之後,它們之間能形成哪些基本構型,這被稱爲蛋白質的二級結構;這些構型之間如何相互靠攏、形成有功能的團塊,這被稱爲蛋白質的三級結構;有的蛋白質還需要幾條多肽鏈之間的相互結合,這被稱爲蛋白質的四級結構。
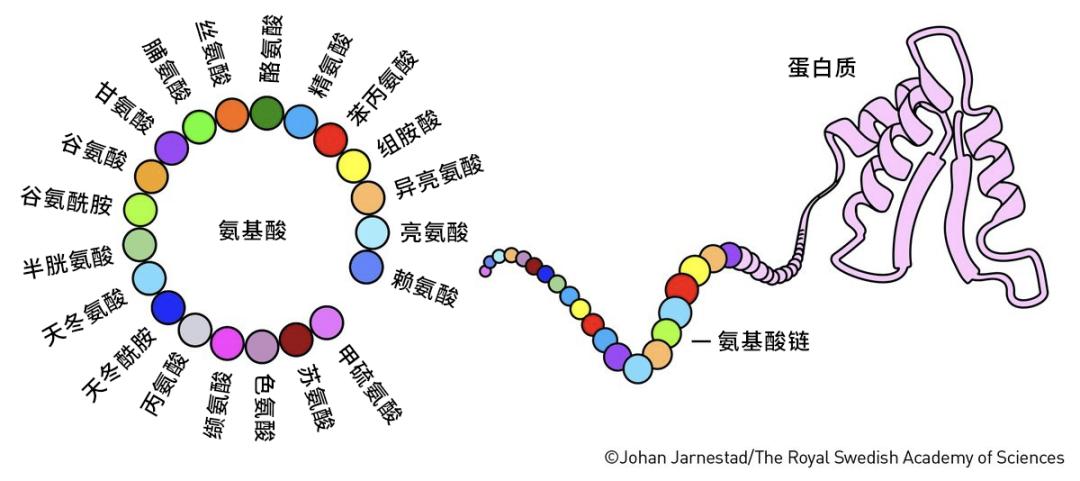
蛋白質的結構 (圖片來源:瑞典皇家科學院)
人們發現,蛋白質的複雜功能,完全依賴於蛋白質的三級和四級結構。蛋白質從線性的多肽鏈形成複雜卻精巧的三維結構,這就是蛋白質摺疊。生物中的每個蛋白質都是極致的平衡大師。它們的形狀、大小、親水性、電荷數量等特性被嚴密地控制在一個最適合的範圍內,從而保證它們各自能夠行使不同的生物學功能。
而這些高級結構能夠維持,又是由於一級結構所決定的。一級結構如果出錯,那麼就會導致所有的高級結構統統錯誤,嚴重情況下會導致整個蛋白質失效。
例如,如果在血紅蛋白的某個位置上,本來親水性的氨基酸突變成了不親水的氨基酸;這個不親水的氨基酸會導致這個血紅蛋白的親水性大大降低,於是它們不是溶解在血液裏,而是互相凝固在一起。凝固在一起的血紅蛋白無法攜帶氧氣,這就是鐮刀形紅細胞貧血病的病因。
因此,蛋白質的一級結構是基礎,而它的高級結構是表象。那麼,我們是否可以從基礎推測表象,或者依據想要實現的表象,而設計出基礎呢?
要了解蛋白質結構的祕密,太難了
一級結構的測定方法早在上世紀七十年代就已經問世。而從上世紀五十年代開始,化學家們就已經能夠根據氨基酸的性質,推測相鄰氨基酸之間的相互作用,以及它們能夠形成的構型,因此人們對二級結構的瞭解也很深入。
此時,人們已經確信,蛋白質的高級結構,完全由其氨基酸序列確定。但這也帶來了一個悖論:氨基酸序列能夠產生的排列可能是一個天文數字,如果細胞把每個可能性都嘗試一遍,那可能要到宇宙毀滅那一天。換句話說,蛋白質是如何快速“選擇”自己該摺疊成什麼結構的呢?其背後的規則是怎樣的呢?
一旦掌握了這個規則,那麼我們就能解決兩個重要問題:一是蛋白質結構預測,二是蛋白質結構設計。
換句話說,一旦掌握了蛋白質摺疊的規則,那麼人類就成爲了主宰蛋白質世界的“神”,我們就可以快速解讀出世界上任何蛋白質的功能,從而清晰地認識生物;也可以任意向生物中添加某種功能的蛋白質,從而定向地改造生物。
這麼重要的規則,其難度當然可想而知。
用樂高的比喻來說,這兩個問題就是:如果給我一堆樂高零件,我能不能預測一下它們能拼成什麼物體;如果讓我去拼一個物體,我能不能徒手畫出它的設計圖,並且判斷哪種設計圖最省時省力。
對於樂高來說,各個小塊之間的相互作用是確定而穩固的,它們互相可以卡住,並且只能從固定的方向卡住。
但是對於氨基酸來說,它們可能以許多類型的力進行相互作用,例如電荷相互作用、疏水作用、氫鍵等。更要命的是,這些氨基酸在空間上可能以任意的角度、方向和距離進行相互作用;而且相互作用的大小、類型都會爲不同的功能來服務,並不是一成不變的。
起初,人們希望通過解構一些有代表性的蛋白質的結構,對蛋白質摺疊規則來個“管中窺豹”。結果窺了好幾百次豹才發現,每次窺到的東西都不一樣,有時候還不能確定窺到的是不是豹子身上的斑點。
畢竟,蛋白質的功能有幾十萬種,結構有幾百萬種,它們內部到底隱藏着多少規則,這已經遠遠超出了人腦的理解能力範圍。
利用人工智能的強項解決問題
然而,這種從大量重複中尋找固定模式,並且引用這一模式來解決實際問題的工作,恰好是人工智能的強項。
人工智能能夠將蛋白質的幾十萬種功能和幾百萬種結構一個個地學習並總結起來,它雖然不能向人類描述其中的摺疊規則,但是卻能夠合理地運用它所發現的規則,從而間接實現蛋白質結構設計和結構預測。
下面就是今年獲獎者們的一些嘗試。
1999年,貝克爾和同事開發了Rosetta計算機程序。這一程序先是學習了當時人類已知的蛋白質數據庫,然後使用蒙特卡洛優化,主要考慮範德華相互作用、氫鍵和溶劑化效應等因素,從而給出幾個可能得方案。
貝克爾用這一程序設計了一個特定功能的蛋白質,程序給出的方案與任何自然界中存在的蛋白質都不同;人們將它合成出來之後,確實發現它能夠行使貝克爾預先設想的生物學功能。這也是人類首次成功設計出複雜功能的蛋白質。
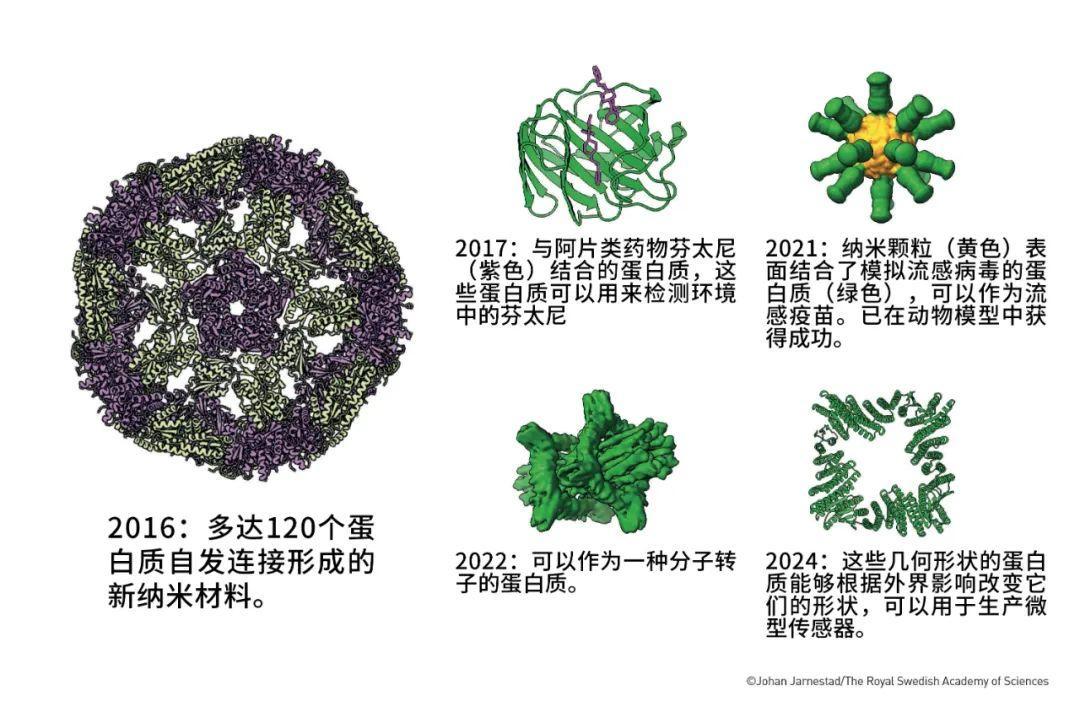
一些利用Rosetta設計的蛋白質(圖片來源:瑞典皇家科學院)
時間到了2018年,哈薩比斯和詹珀的Deepmind公司,開發了基於卷積神經網絡的人工智能AlphaGo,通過學習了上萬億盤圍棋之後掌握的圍棋規則,徹底擊敗了人類;於此同時,Deepmind公司將這種快速學習的人工智能應用於蛋白質結構預測領域,開發了AlphaFold,預測成功率達到了60%。
到了2020年,AlphaFold的升級版AlphaFold2的預測結果已經可以做到與實驗測量結果幾乎沒有誤差,也就是說只要告訴它蛋白質是由哪些氨基酸構成的,那麼它就會告訴你這個蛋白質的結構和功能是怎樣的。而2024年發佈的AlphaFold3雖然精確度變化不大,但具有了一定的通用性,不再侷限於蛋白質,還可以用於其他生物高分子與小分子配體、高分子修飾,以及蛋白質和它們的複合物結構。

Alphafold2基於神經網絡的深度學習模型 (圖片來源:“返樸”公衆號)
結語
可以說,貝克爾、哈薩比斯和詹珀的工作開闢了生化和生物學研究的新時代,我們現在可以用以前無法想象的方式預測和設計蛋白質結構。
當然,如果沒有結構生物學家的努力,上述進展不可能實現。他們爲蛋白質數據庫提供了大量實驗確定的蛋白質結構。這些數據是數十年蛋白質結構測定研究的結果,爲今年獲獎者在蛋白質設計和結構預測方面取得的決定性突破奠定了基礎。
有人也許會問“人工智能這麼強大了,結構生物學家是否會失業?”其實,就像ChatGPT沒有讓寫手失業一樣,人工智能在蛋白質設計和結構預測方面還有很多問題有待解決(比如對複合體、柔性區的預測等等)。對於結構生物學家來說,“獲得結構”只是研究的手段,理解生命,做出生物學發現纔是目的。正如顏寧院士所說,“如何能夠理解我們細胞裏各個分子的動態變化,是我們目前面臨的最大挑戰之一”。
作者:牧心